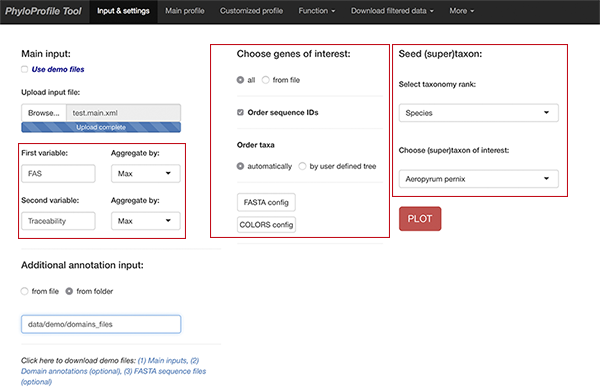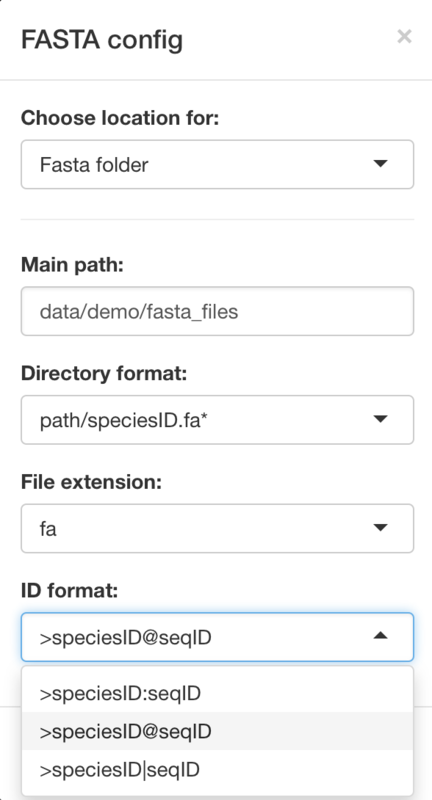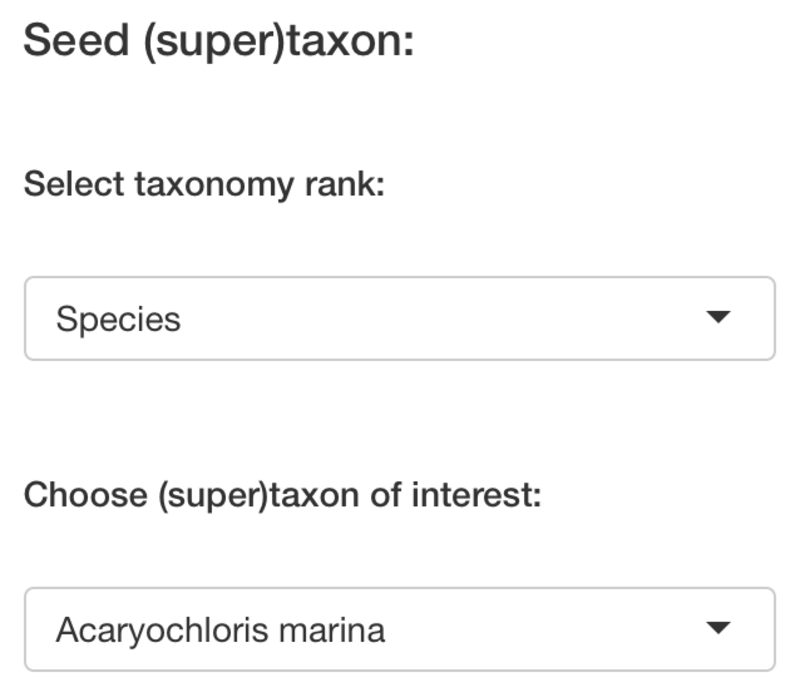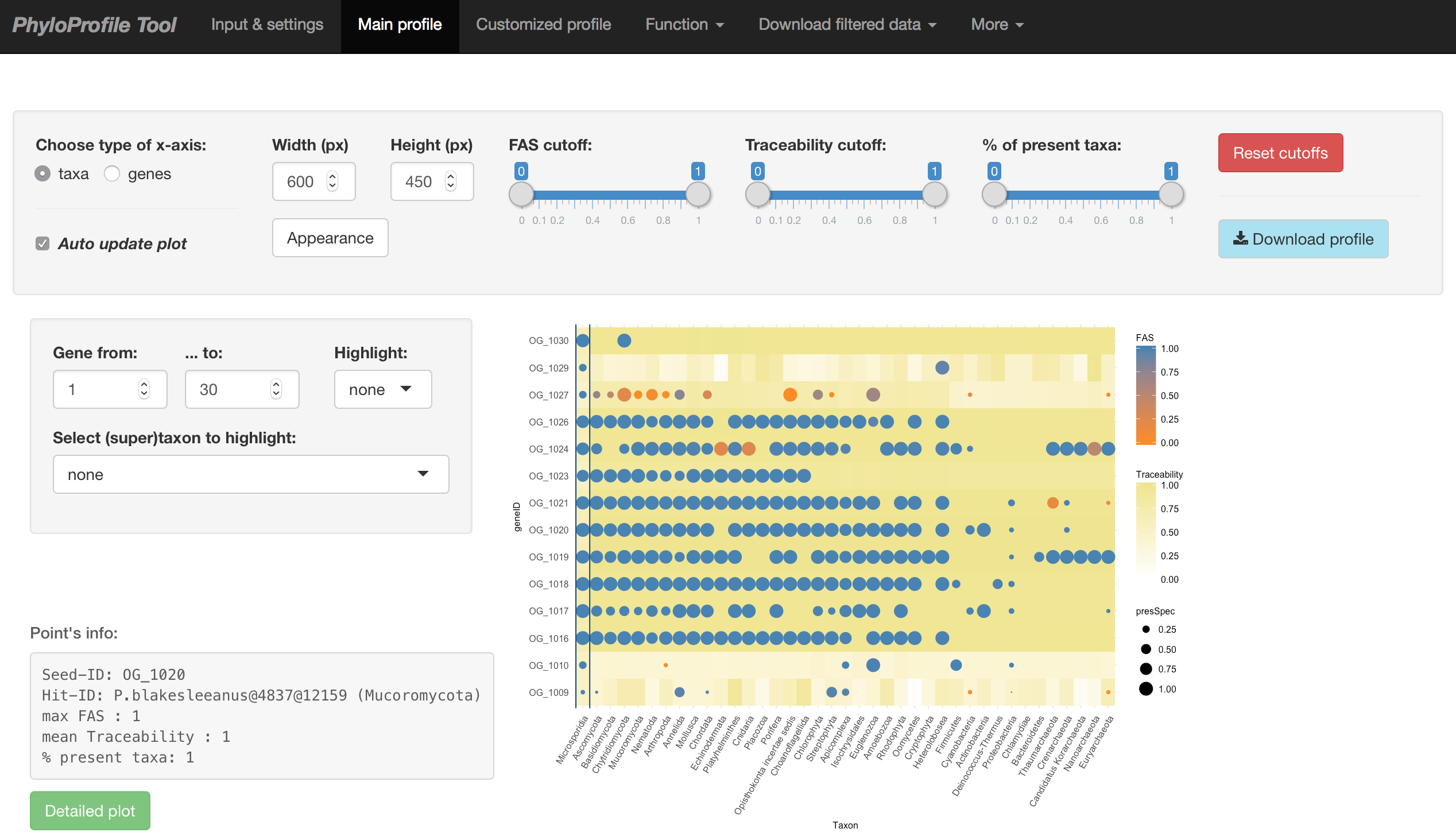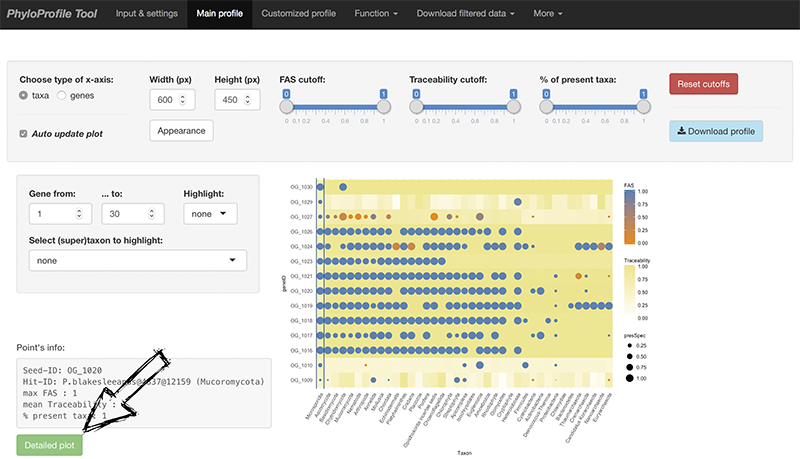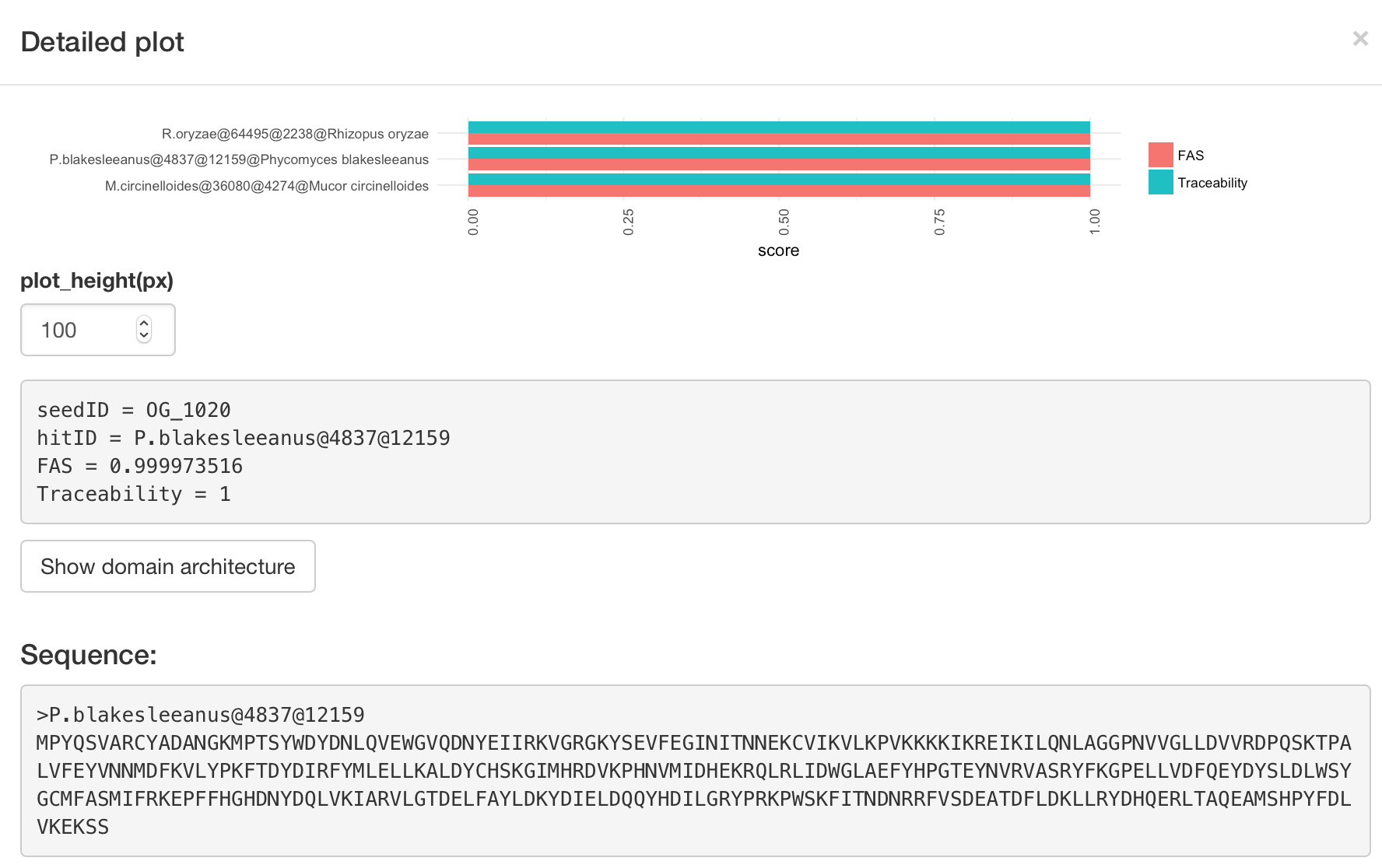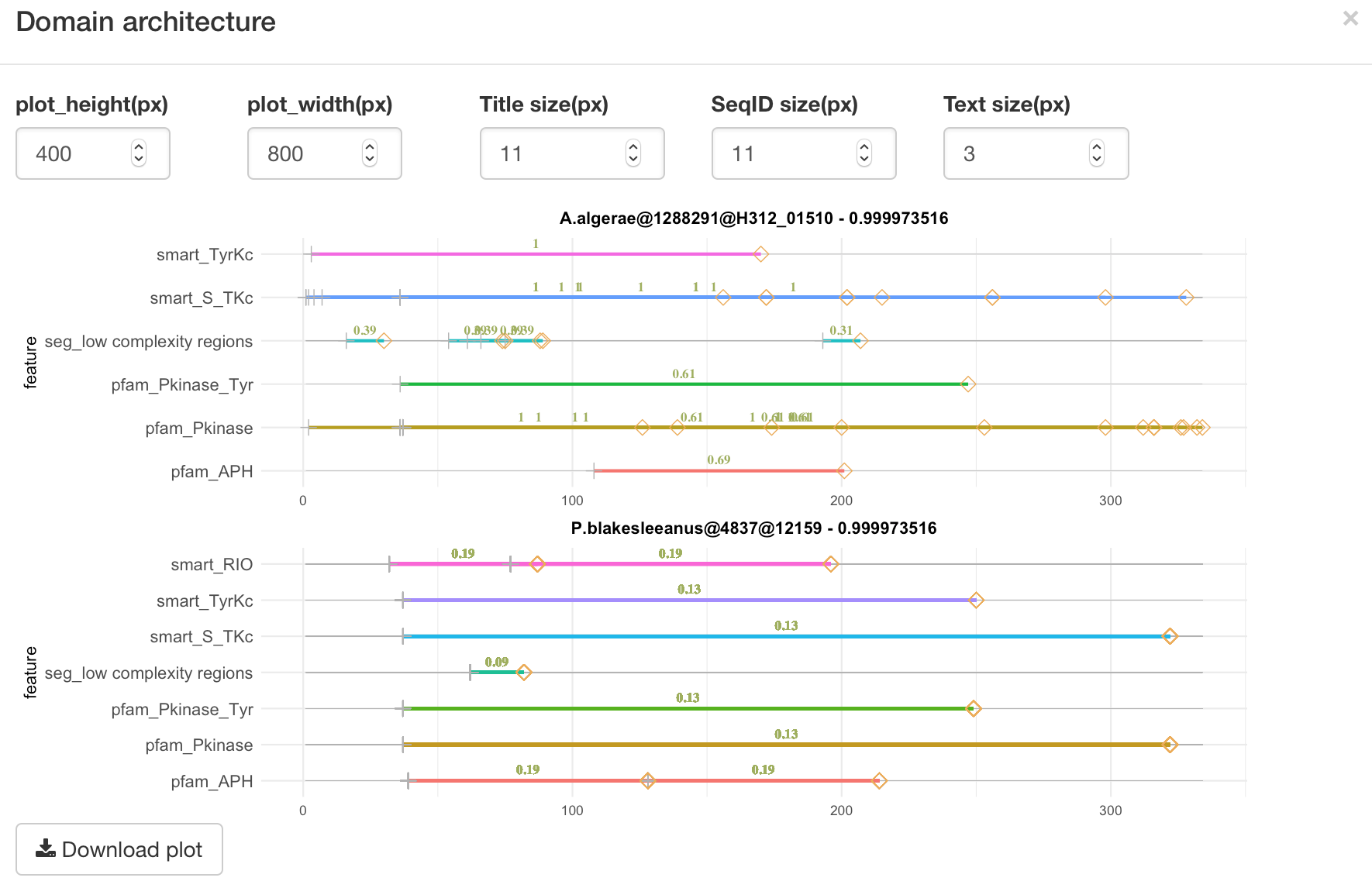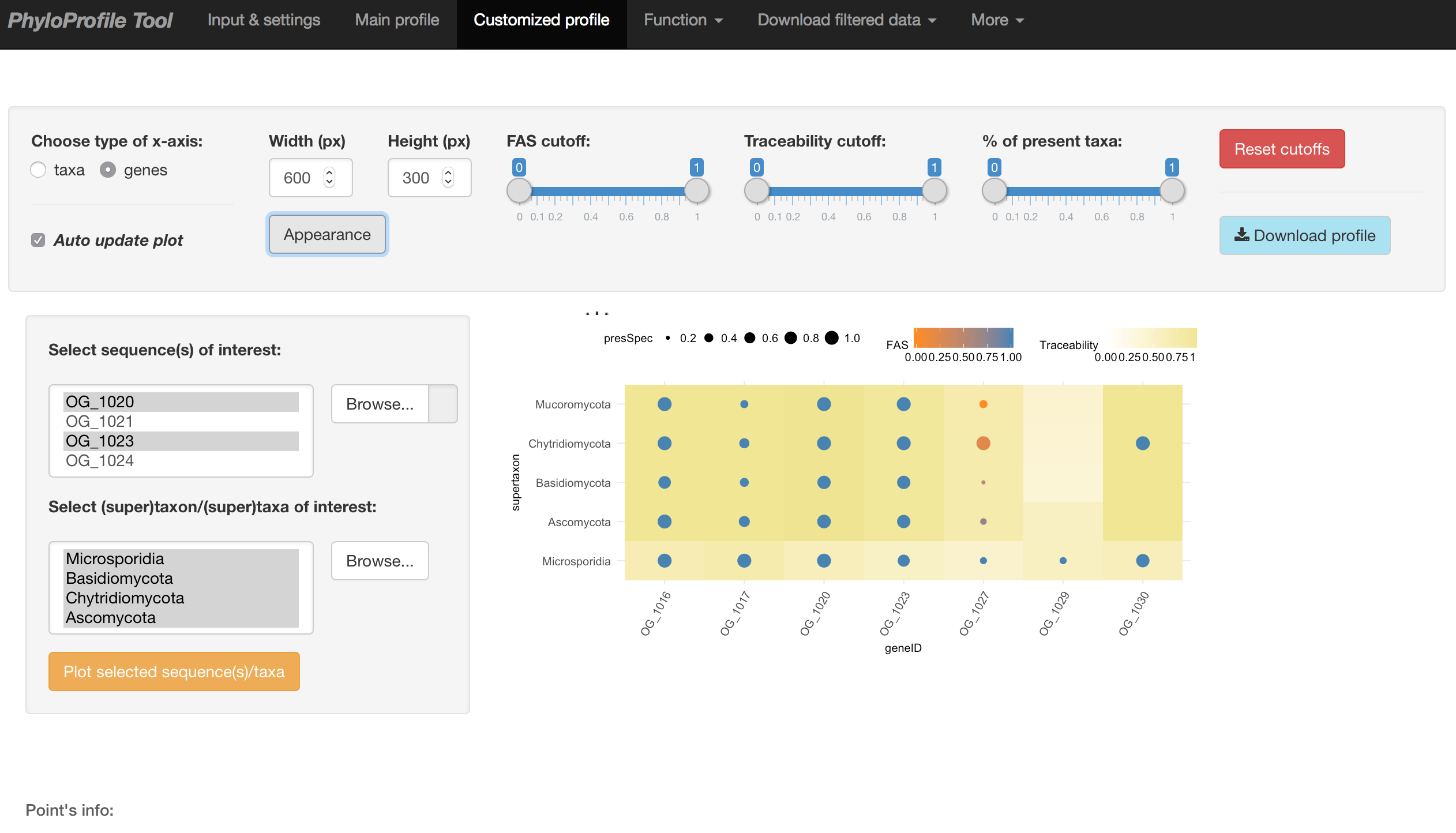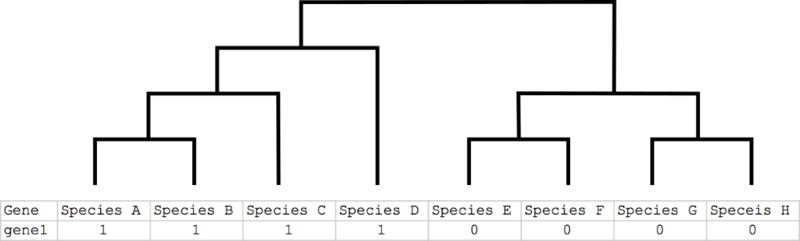

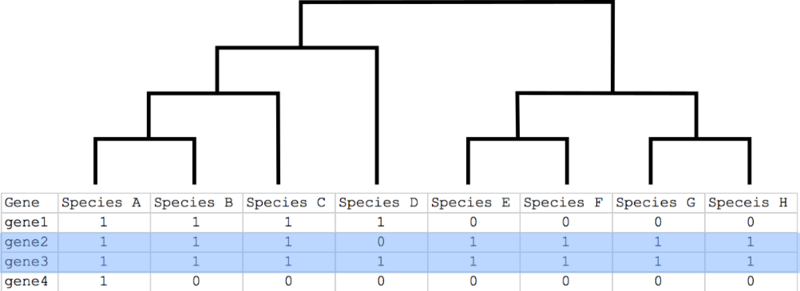
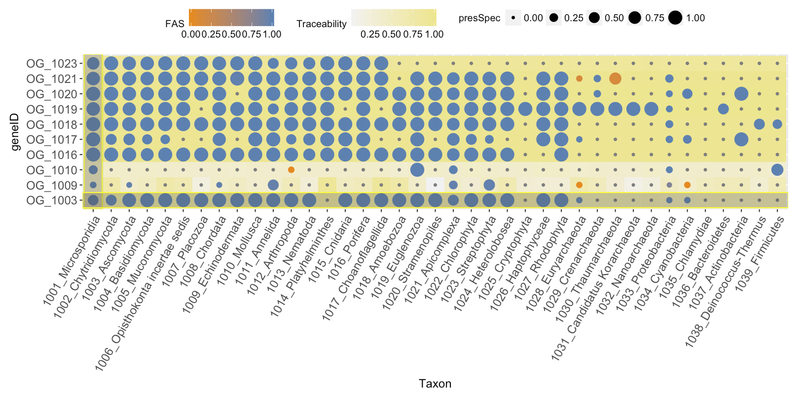
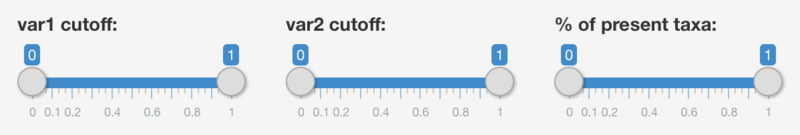
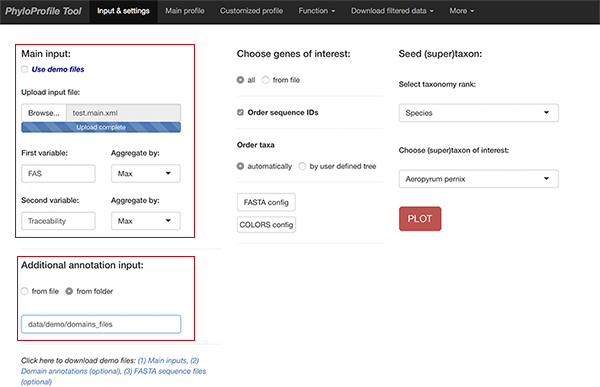
Main input
Required information
- for basic phylogenetic profile:
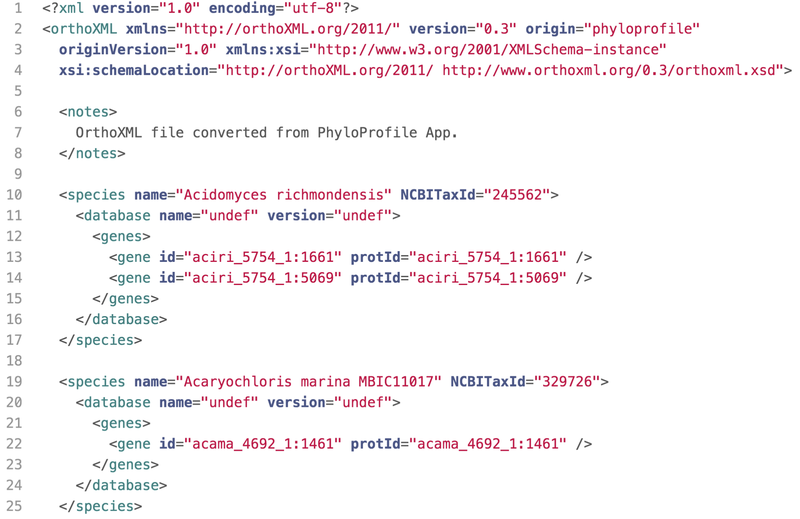
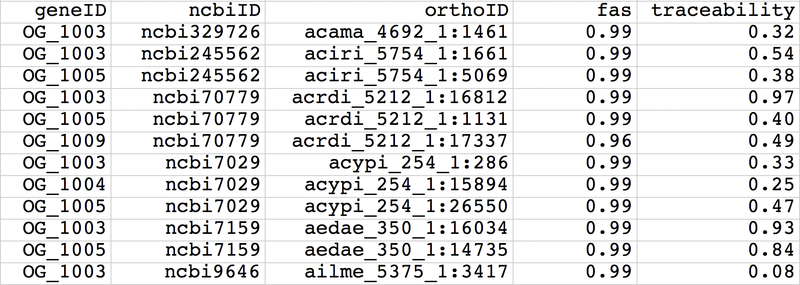
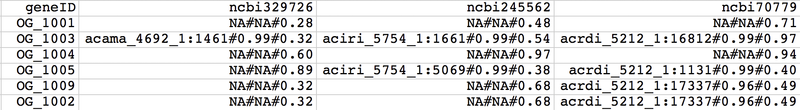
- Gene ID of seed protein or ortholog group ID - geneID
- Taxonomy ID of species having orthologs (ncbi+taxonID, e.g. ncbi7029, ncbi3702) - ncbiID (*)
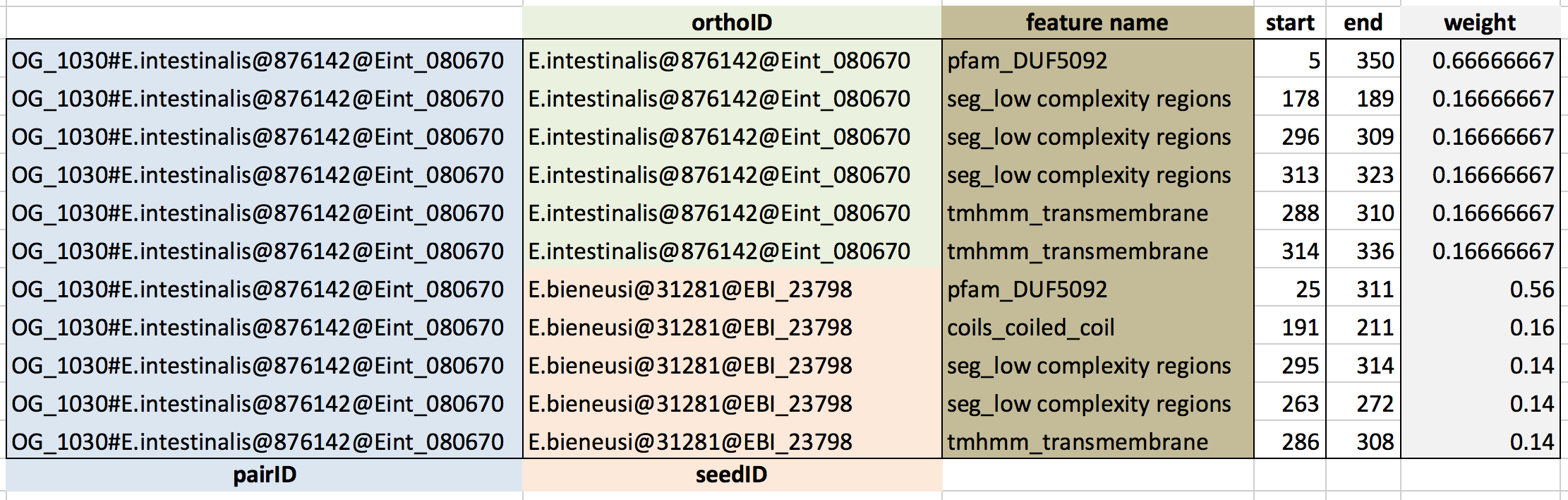
- Ortholog ID - orthoID
- for additional information layers (optionally):
- Value for first additional information layer - var1
- Value for second additional information layer - var2
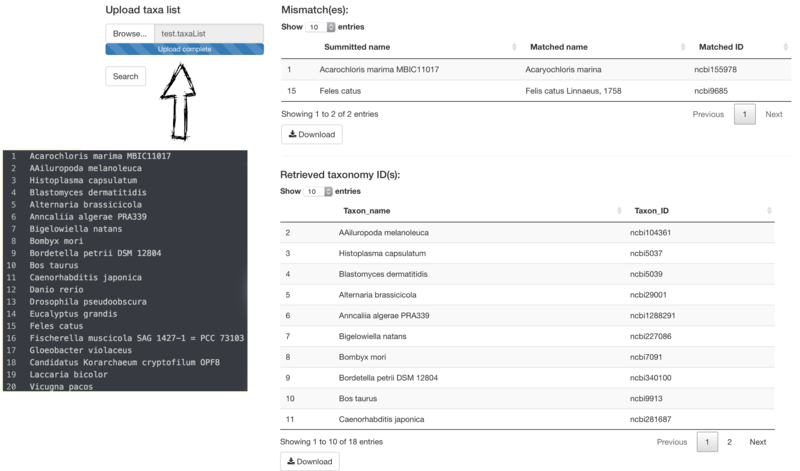
(*) PhyloProfile requires taxonomy IDs in all of input files.
PhyloProfile is provided with a function for searching taxon IDs from a list of taxon names.